Here I'll share some experiences for sequencing samples by RNA. Most of them will be useful for other techniques and also for single cell sequencing (or scRNA-seq). Here I'm assuming that you do a short-sequence technique from Illumina or similar companies. Also I'll provide some tips to make easier the life of the bioinformatician that will analyse the data.
Don't expect to find how many reads for sample you need or which design is better or which machine. Neither I will talk about prices or provide a list of companies and scientific platforms to look for. I won't cover how to analyse the data, or compare pipelines to process RNA-seq. Hope this post is more practical than all that but less technical.
You carried out your experiments and have already extracted the RNA from patients, mice, rats, cell cultures or whatever are you studying. You have them named with sensible names for your experiment like A+B-, 4789/Diff, 256-w046 and usually stored in your notebook and probably also in a spreadsheet (Remember to annotate too where did you store them of the freezer). Also be consistent with your naming scheme, don't store some samples as 256-w046 others as 257-w46 and others as 256-W046, this will make it harder to relate the samples of your experiments to the sequencing samples and mistakes might happen when matching between the names of the samples.

Before going further you need to measure the quality of the material, in RNA by looking at the RIN, concentration and volume you have. Platforms and companies usually require a certain volume at certain concentration or X amount of RNA and a good quality to process the samples.
Next come sending the samples you selected for sequencing. As any experiment it is recommended to have some quality controls so I recommend to send also some water samples to verify that there isn't any problem with the process (contamination from other samples, degradation...) . You should also try to have some replicates of the samples, there is ample evidence of the importance of replicates on sequencing 1, 2. I know sometimes is difficult to have biological replicates but at least try to have technical replicates if you have enough volume.
Depending of the machines you'll use and the number of samples to sequence they will be sequenced on several batches. Make sure to account for that when sending the plates and avoid a batch effect. Either block correctly or randomize the samples of each batch. For this purpose I created a tool to randomize the samples and minimize the batch effect: experDesign.
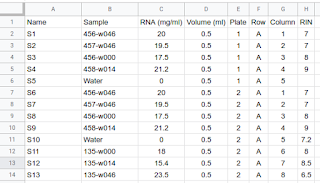
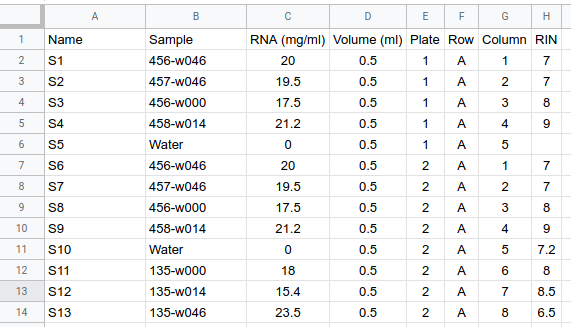
I suggest creating a new spreadsheet file with all the information they required, RIN, concentration, position on the plate, number of plate if you make a big batch... the name of the sample (the original one) and the new name you provide to the sequencing people that should be easy to write and short. This new name is necessary if you include replicates and water as control as you need to be able to differentiate between different replicates of the same sample. I suggest use S1, S2, S3, S4, .... This file will be quite useful later on, but the sequencing platform might require an specific format or files and might provide you with templates and examples.
Next is how you send the samples, physically. You'll need to make sure they are not damaged along the way, so you'll need send them with dry ice and have the box replenish if you send it far away. If it is on the same centre you'll might just need to use ice.
Once the samples arrive you'll need to wait their quality check in case the samples degradated on the trip, again RIN, concentration, volume. will be measured. If some samples do not meet their threshold they might ask you to send more volume or see if you can send samples with higher quality. This process might be repeated several times, so you might end up sending several times the same sample.
With the samples that meet their standards they will prepare the library and sequence them. This is the most expensive part. This might take between 1 or 3 months.
They might have created you an account for you to download the data from their servers, and provided with credentials and how to log in there. You might receive an email once each batch is sequenced or you might need to check once in a while if there is something to download. The files will usually be .fastq.gz. If you are using two-way sequencing (recommended) you might have two files for each sample (S1_R1.fastq.gz and S1_R2.fastq.gz).
The files are at least 1 month on this servers, so make sure to check at most every two weeks. Also take into account that it might take some days to download the data. If the batch is big enough it could be faster just to send the disk by mail than just download it.

Once the files are securely stored you need to check them:
Once all the samples pass these quality controls you can start with the pipeline you need. This will depend on the type of question you want to answer and the sequencing technique applied.
Don't expect to find how many reads for sample you need or which design is better or which machine. Neither I will talk about prices or provide a list of companies and scientific platforms to look for. I won't cover how to analyse the data, or compare pipelines to process RNA-seq. Hope this post is more practical than all that but less technical.
Before sending: Get ready
You carried out your experiments and have already extracted the RNA from patients, mice, rats, cell cultures or whatever are you studying. You have them named with sensible names for your experiment like A+B-, 4789/Diff, 256-w046 and usually stored in your notebook and probably also in a spreadsheet (Remember to annotate too where did you store them of the freezer). Also be consistent with your naming scheme, don't store some samples as 256-w046 others as 257-w46 and others as 256-W046, this will make it harder to relate the samples of your experiments to the sequencing samples and mistakes might happen when matching between the names of the samples.

Before going further you need to measure the quality of the material, in RNA by looking at the RIN, concentration and volume you have. Platforms and companies usually require a certain volume at certain concentration or X amount of RNA and a good quality to process the samples.
Next come sending the samples you selected for sequencing. As any experiment it is recommended to have some quality controls so I recommend to send also some water samples to verify that there isn't any problem with the process (contamination from other samples, degradation...) . You should also try to have some replicates of the samples, there is ample evidence of the importance of replicates on sequencing 1, 2. I know sometimes is difficult to have biological replicates but at least try to have technical replicates if you have enough volume.
Depending of the machines you'll use and the number of samples to sequence they will be sequenced on several batches. Make sure to account for that when sending the plates and avoid a batch effect. Either block correctly or randomize the samples of each batch. For this purpose I created a tool to randomize the samples and minimize the batch effect: experDesign.
I suggest creating a new spreadsheet file with all the information they required, RIN, concentration, position on the plate, number of plate if you make a big batch... the name of the sample (the original one) and the new name you provide to the sequencing people that should be easy to write and short. This new name is necessary if you include replicates and water as control as you need to be able to differentiate between different replicates of the same sample. I suggest use S1, S2, S3, S4, .... This file will be quite useful later on, but the sequencing platform might require an specific format or files and might provide you with templates and examples.
Next is how you send the samples, physically. You'll need to make sure they are not damaged along the way, so you'll need send them with dry ice and have the box replenish if you send it far away. If it is on the same centre you'll might just need to use ice.
What to expect of the sequencing centre:
Once the samples arrive you'll need to wait their quality check in case the samples degradated on the trip, again RIN, concentration, volume. will be measured. If some samples do not meet their threshold they might ask you to send more volume or see if you can send samples with higher quality. This process might be repeated several times, so you might end up sending several times the same sample.
With the samples that meet their standards they will prepare the library and sequence them. This is the most expensive part. This might take between 1 or 3 months.
They might have created you an account for you to download the data from their servers, and provided with credentials and how to log in there. You might receive an email once each batch is sequenced or you might need to check once in a while if there is something to download. The files will usually be .fastq.gz. If you are using two-way sequencing (recommended) you might have two files for each sample (S1_R1.fastq.gz and S1_R2.fastq.gz).
The files are at least 1 month on this servers, so make sure to check at most every two weeks. Also take into account that it might take some days to download the data. If the batch is big enough it could be faster just to send the disk by mail than just download it.
Samples are sequenced: now what?
As the samples are provided I recommend that you download and store in at least two separate devices or disks. Check that you downloaded them correctly using md5sum. You might want them all files in a single folder or a folder for each batch.Once the files are securely stored you need to check them:
- First check that the amount of files is correct. Double the number of samples. Also sometimes with the first sequencing they don't reach the number of reads you paid for so some samples might be run in another batch and you might end up with 4 files or more for a single sample.
- Check that the files are correctly formatted. Once we got a truncated fastq file and only realized it when we uploaded to a public data repository. I use FastQValidator for that since then.
- Check that the quality is correct. Usually done with FastaQC, some facilities even provide this quality report themselves. At this stage if you find something wrong the only options are remove the sample or send it again and make a new library for it.
Once all the samples pass these quality controls you can start with the pipeline you need. This will depend on the type of question you want to answer and the sequencing technique applied.